

Señor Presidente de la Academia Nacional de Medicina
Señores Individuos de Número, Miembros Correspondientes e Invitados de Cortesía y Asociados
Doctor José Guevara Iribarren
Familiares y amigos del Doctor José Guevara Iribarren
Señoras y señores
Con gran complacencia, íntima satisfacción y sinceridad, expreso mi agradecimiento al señor Presidente, Dr. Claudio Aoún S. y demás miembros de la Junta Directiva de esta honorable y docta Academia por haberme distinguido con el honor de seleccionarme para hacer el juicio crítico reglamentario sobre el trabajo de incorporación de mi dilecto amigo, el Doctor José María Guevara Iribarren, ¨Desarrollo del Estudio de las Hemoglobinas Anormales en Venezuela¨, quien por propia densidad intelectual viene a ocupar con el beneplácito de todos los miembros de esta corporación el Sillón XXX, y a quien quedo reconocido por su benevolencia y aquiescencia.
Trataré de presentar, más que criticar, el valor y alcance de su trabajo, habida cuenta la liviandad de mis conocimientos en una materia a la cual nuestro recipiendario ha dedicado tantos años y desvelos. Pido además a mis estimados compañeros académicos, colegas invitados y al público presente, su benevolencia por la inusual forma en la que comenzaré mi discurso, por ahora, alejado del rigor científico y más emparentado con la urdimbre espiritual del ser humano, la segregación racial y el sufrimiento:
–» ¿Qué hace este niño en un hospital de adultos?», me pregunté cuando le vi pasmado, enteco y flaquito, espalditendido, febricitante y sudoroso, con su abultada barriga y sus piernitas tan delgadas como tacos de billar, desmesuradas a la altura de los tobillos donde, como nudos en el tronco de un árbol añoso, se abrían sendos cráteres ulcerosos, dos volcanes de sanioso fondo en dolorosa actividad… ¡Parecía embuste: contábamos ambos 23 años, pero nos había tocado suertes tan diferentes….! Yo le miré con aire de clínico en ciernes, imitando a mis mayores, tratando de arrancarle un diagnóstico al vistazo, aquel d΄emblé de los afrancesados; él contestó mi mirada de pseudocientífico en vías de mecanización, con una cálida y adolorida sonrisa, mostrándome mucho más que sus marfileños dientes, apenas al descubierto por la brecha que sus gruesos labios entreabiertos dejaban, en notorio contraste con la negritud de su tez. Desde entonces y hasta ahora, por una cuarentena de años, el negrito Güilian, ha sido mi paciente. De común vestido a lo pachuco, con su invariable flucecito blanco y sus botas de tacón cubano, orgulloso les dice a mis alumnos, que él me conoció cuando yo era un simple interno con cara de bebé, pero disfrazado de más grande con mi bigotico a lo Jorge Negrete. ¡Qué cosa!, antes parecía mi hermano mayor, y ahora parezco yo su abuelo. Cada vez que reúne algún dinero para pasaje y bastimento, viene desde Barlovento sin cita a visitarme, precisamente desde el lugar donde negros de la Costa de Marfil y de la Costa de Oro eran traídos al puerto más importante de esta región llamado San Jorge de Mina; así que los negros que allí llegaron, fueron conocidos como «mina». Todavía hay un tambor conocido con el mismo nombre en el Barlovento venezolano.
Entonces, me resultó difícil comprender el porqué de los privilegios con que la vida me había premiado, siendo que a él… se los había negado todos. Me dio pues, por hacer odiosas comparaciones: Nacimos el mismo mes del mismo año y yo le llevo cinco días. El doctor Ramón Cifuentes parteaba mi difícil salida en posición podálica un domingo 1º de mayo, cuando a él, el viernes siguiente, una vecina medio sabida, hacía que atendía a su mamá, la única figura parental de que supo Güilian… el sempiterno y ausente padrote criollo, la montó tres años seguidos, le secó sus turgentes senos y le robó la tonicidad al fondo de sus entrepiernas entre noches de borracheras, imprecaciones y maltratos; y así, sin dejarle ver un solo período, le sembró tres negritos, todos ellos con mezcla de la tara genética que ambos traían a cuestas… No accedió a escuela alguna, y desde que supo, fue un “habitué” del Hospital de Niños de Caracas, desde donde con tristeza por dejar sus médicos atrás, se vino al Vargas, donde el destino me hizo adoptarlo como paciente. Dígame yo, a quién nada me había faltado: Hasta sarna para rascarme, que habría de pegarme un hippy melenudo y malhablado por allí, en tiempos de Woodstock y su estridente música… Poco él, mucho yo. ¿Habría sido yo consecuente con las prebendas económicas, sociales y profesionales con las que el destino, por pura buena suerte, me había hartado…?
¨Veamos – cavilé-, negrito jipato con fiebre, severo dolor abdominal y de las coyunturas, ojos tatuados del amaranto ictérico y úlceras en las piernas…¨ ¡Una adivinanza médica más fácil de acertar que echarle un tiro al suelo!, tan sencillo como descubrir cuál es ese animalito cuadrúpedo, de largo rabo, puntiagudas orejas, elegante bigote, que hace… ¡ñau! ¡Anemia drepanocítica o de células falciformes! ¨ dije enfático en mí interior.
Alguna vez escribí que el estudio de las hemoglobinopatías en Venezuela traía consigo, por una parte, el recuerdo de la llegada a nuestras tierras de emigrantes del Mediterráneo; sangre nueva, industriosa, simple e ingenua, pero no siempre de noble constitución; y por la otra, de mareados y tristes tiempos de negros, y luego zambos y mulatos, de esclavos asentados en la franja litoral, ¨barcos con cargamentos negreros¨ atiborrados de bantúes, olofes y gangás, procedentes de mercados de esclavos enclavados en la Costa Occidental del África Congo-angoleña, trasunto de minas y tambores culo´epulla, expurgación de espíritus malignos, fetiches y tizas rituales, restallido de látigos de negreros, reminiscencias de vividores sólo del presente, sufrimiento inmenso, ¨buenos pa´cargá¨, negación del derecho humano, desarraigo, páginas de la patología médica afroamericana, muerte temprana, pero también… el aromoso e ingenuo recuerdo de las venerables madres negras de El Libertador, Hipolita Bolívar la nodriza y Matea Bolívar, el aya…
 Con ellos, fumando “con la candela pa’dentro”, se vino coleada la tara drepanocítica o falciforme, que etimológicamente expresa la deformidad del glóbulo rojo en forma de hoz o semiluna turca. Una pifia genética, donde el glóbulo rojo normal, redondito, sonrosado y maleable para poder deslizarse por intricados vericuetos de delgadísimos capilares hasta la célula recóndita llevándole su carga de oxígeno, adquiere la forma de una hoz, rígida y chata, incapaz de acarrear el gas vital hasta la intimidad tisular, porque entrelazándose unos con los otros, se atoran mecánicamente, produciéndose “trancas” fenomenales en uno y mil capilares, conduciendo a mal funcionamiento de todos los órganos, aparatos y sistemas. Un único cambio en la monótona secuencia de aminoácidos en la cadena beta de la hemoglobina -una de las cuatro cadenas polipeptídicas que la constituyen-, valina por ácido glutámico, una minúscula sustitución, produce una hemoglobina anormal llamada S, que al encontrarse en un medio pobre en oxígeno deforma al glóbulo rojo transformándolo en inútil guadaña. Cuando la hemoglobina se retuerce y se apelotonan los glóbulos rojos en la microcirculación, hace su aparición la terrible crisis drepanocítica con su séquito de destrucción de los pocos glóbulos rojos existentes y anemia agravada, infartos orgánicos y óseos múltiples, muerte de tejidos y dolores insoportables, infecciones recurrentes en el sujeto asplénico –ausencia del bazo-. Si ambos progenitores poseen esta hemoglobina S, al hijo, como al Güilian, se le llama homocigoto y llevará a cuestas toda su vida y sufrirá la dolencia anemizante… ¨.
Con ellos, fumando “con la candela pa’dentro”, se vino coleada la tara drepanocítica o falciforme, que etimológicamente expresa la deformidad del glóbulo rojo en forma de hoz o semiluna turca. Una pifia genética, donde el glóbulo rojo normal, redondito, sonrosado y maleable para poder deslizarse por intricados vericuetos de delgadísimos capilares hasta la célula recóndita llevándole su carga de oxígeno, adquiere la forma de una hoz, rígida y chata, incapaz de acarrear el gas vital hasta la intimidad tisular, porque entrelazándose unos con los otros, se atoran mecánicamente, produciéndose “trancas” fenomenales en uno y mil capilares, conduciendo a mal funcionamiento de todos los órganos, aparatos y sistemas. Un único cambio en la monótona secuencia de aminoácidos en la cadena beta de la hemoglobina -una de las cuatro cadenas polipeptídicas que la constituyen-, valina por ácido glutámico, una minúscula sustitución, produce una hemoglobina anormal llamada S, que al encontrarse en un medio pobre en oxígeno deforma al glóbulo rojo transformándolo en inútil guadaña. Cuando la hemoglobina se retuerce y se apelotonan los glóbulos rojos en la microcirculación, hace su aparición la terrible crisis drepanocítica con su séquito de destrucción de los pocos glóbulos rojos existentes y anemia agravada, infartos orgánicos y óseos múltiples, muerte de tejidos y dolores insoportables, infecciones recurrentes en el sujeto asplénico –ausencia del bazo-. Si ambos progenitores poseen esta hemoglobina S, al hijo, como al Güilian, se le llama homocigoto y llevará a cuestas toda su vida y sufrirá la dolencia anemizante… ¨.

Para entrar en materia tal vez sea razonable resumir en forma sucinta las manifestaciones clínicas de las hemoglobinopatías congénitas. La más frecuente entre nosotros, la anemia de células falciformes o drepanocítica, fue descrita por James Henrick en 1910; una enfermedad genética autosómica recesiva resultado de la sustitución de adenina por timina en el gen de la globina beta, ubicado en el cromosoma 11, lo que conduce a una mutación de ácido glutámico por valina en la posición 6 de la cadena polipeptídica de globina beta y a la producción de una hemoglobina funcionalmente defectuosa, la hemoglobina S. Debido al cambio de ese aminoácido, las moléculas de hemoglobina se agregan formando fibras y dándole al hematíe forma de hoz. La transformación del eritrocito redondo y maleable se produce cuando no transporta oxígeno, pues en presencia de oxihemoglobina, el glóbulo mantiene la forma clásica bicóncava.

Así pues, los principales problemas acarreados por este cambio de forma se deben a la tendencia de los eritrocitos a adoptar una morfología falciforme y a bloquear los capilares cuando la tensión de oxígeno es baja (Figura 1). Los pacientes portadores de una anemia clásica de células falciformes manifiestan variadas anormalidades, incluyendo anemia crónica con hemoglobinemia de alrededor de 8.0 g/dL, infartos óseos, esclerosis ósea (vértebras en ¨boca de pescado¨), necrosis aséptica de la cabeza femoral, isquemia visceral, disnea por infartos pulmonares, aumento de su susceptibilidad a infecciones, particularmente salmonelosis causadas por disfunción del sistema retículo endotelial.
En los niños, los eritrocitos falciformes tienden a quedar atrapados en el bazo, lo cual ocasiona un serio riesgo de muerte antes de los siete años por crisis súbitas de anemia profunda asociada a la rápida esplenomegalia, o por el hipoesplenismo, que permite que se produzcan infecciones muy graves. Entre los 6 y los 18 meses de vida, los niños afectados suelen presentar tumefacción dolorosa de las manos o los pies, el llamado síndrome mano-pie.
Los supervivientes también pueden sufrir crisis dolorosas graves, recurrentes e impredecibles. El dolor puede ser agudo o crónico; pero este último, más frecuente, puede ser brusco y suele ser de moderado a severo. Su duración es de entre horas a días, pudiendo durar hasta semanas o meses. Durante las crisis se afectan los pulmones, el abdomen y las articulaciones constituyendo una emergencia médica. Adicionalmente, la crisis puede presentarse como un ¨síndrome torácico agudo¨, tipificado por neumonía o infarto pulmonar; necrosis articular; priapismo o insuficiencia renal.
En la mayoría de los pacientes es posible reducir la incidencia de complicaciones con medidas de protección simples, tales como la administración profiláctica de penicilina en la infancia, evitación de la deshidratación y del calor o el frío excesivos, o la referencia lo más rápido posible con un centro especializado, recomendándose inmunizar contra el neumococo (PCV-7) y el tratamiento con hidroxiurea (Hydrea®) por su capacidad para aumentar la hemoglobina fetal que reduce la cantidad de glóbulos rojos deformados y la frecuencia de las crisis.
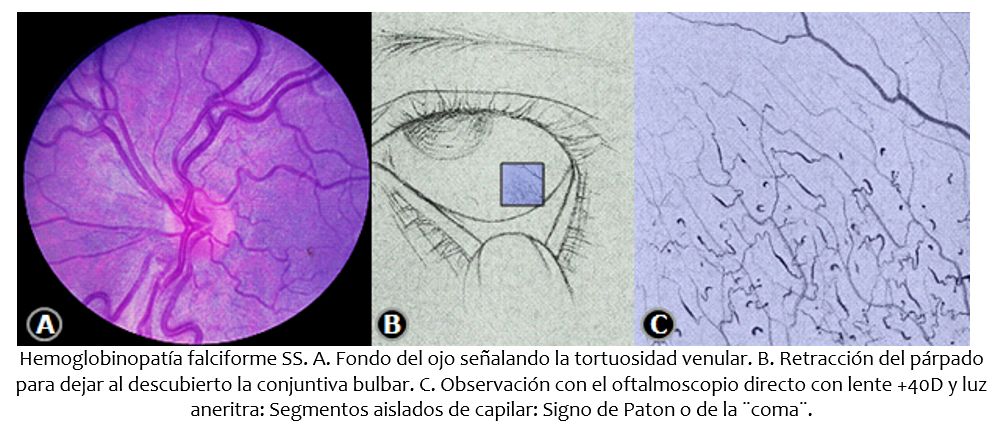
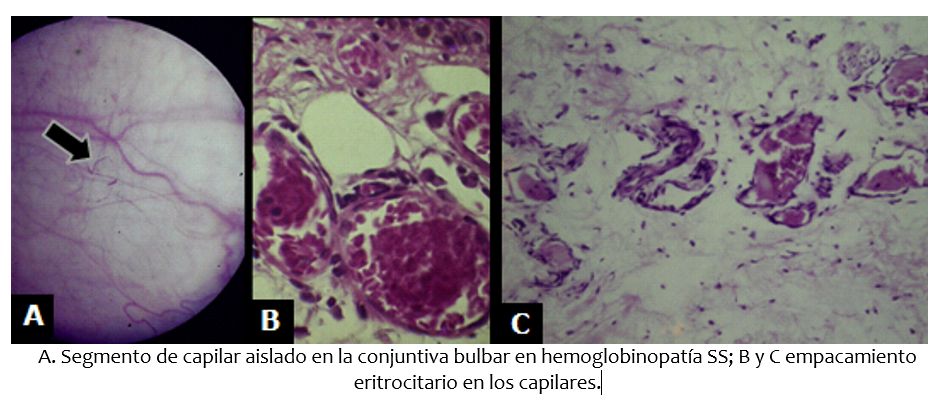
A más de ser interesantes las alteraciones oculares presentes en la drepanocitosis, son importantes porque permiten facilitar el diagnóstico a la cabecera del paciente, y, además, por la morbilidad visual que son capaces de producir. Con relación a la forma homocigota SS, puede decirse que la retina es afectada en un 20% de los casos, en tanto que en la variante SC es del 80%; en la mitad de los primeros se observa distensión y tortuosidad de las vénulas del fondo ocular (Figuras 3 y 5); no sucede lo mismo al observar la conjuntiva bulbar, venero diagnóstico. En 1961 David Paton (1,2) comunicó que la observación biomicroscópica con lámpara de hendidura, permitía apreciar múltiples, cortos y aislados segmentos de capilar separados de la malla capilar simulando comas, tirabuzones o puntos de admiración, y en los cuales el cabo aferente y eferente se encontraban exangües. Su presencia, según el autor, era casi patognomónica de una forma homocigota de drepanocitosis (SS), describiéndolo en un 96% de sus casos; por su parte, en enfermos con hemoglobinopatía SC, su ocurrencia era del 80% (Figura 3). Posteriormente, Comer Fred en 1964 (3), lo puso al alcance del clínico al sugerir su observación mediante el empleo del oftalmoscopio directo, interponiendo una lente de +40 dioptrías que actúa como una lupa de 10X. En 1968 Muci-Mendoza y cols. (4), sugirió interponer el filtro verde incorporado al instrumento para hacer la observación con luz aneritra y así destacar mejor los segmentos anormales; igualmente comunicó sus hallazgos en biopsias conjuntivales en 7 pacientes, tipificados por dilatación sacular de los capilares, proliferación endotelial, empacamiento eritrocitario y células falciformes (Figura 4).
Curiosamente, la HbSC tiene mayor potencial para producir retinopatías isquémicas severas que pueden llegar a la ceguera por desprendimiento retinal. Según Goldberg (5), la retinopatía proliferativa drepanocítica ocurre hacia la tercera década de la vida y se clasifica en 5 estadios (1). Oclusiones arteriolares periféricas que dejan grandes áreas de ausencia de perfusión capilar, mejor apreciada mediante la angiografía fluoresceínica; (2). Anastomosis o cortocircuitos arteriolo-venulares, así que la sangre arterial es derivada a la circulación venosa. Estas comunicaciones se aprecian en el límite de retina perfundida y no perfundida; (3). Proliferación neovascular en forma del alga gorgonia marina (¨sea fan¨); (4). Hemorragias vítreas; y (5). Desprendimiento de la retina (Figura 5).
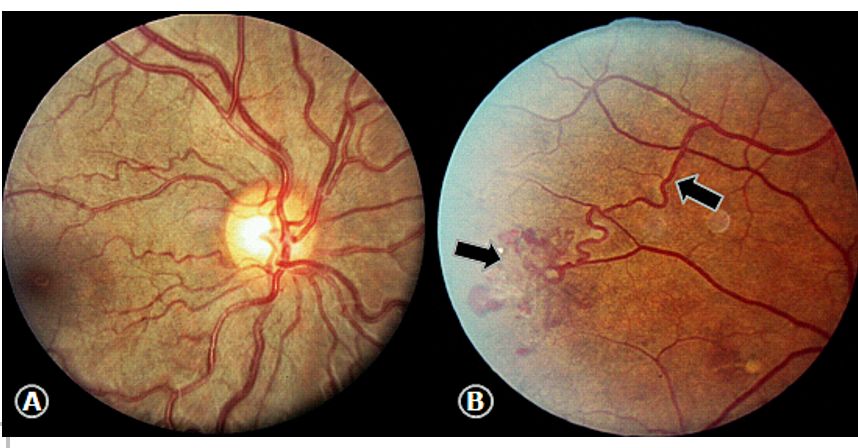
Fondo del ojo anemia falciforme. A. Homocigoto SS discreta tortuosidad vascular.
B. Hemoglobinopatía SC: Cortocircuito arteriolo-venular y neovascularización (¨sea fan¨).
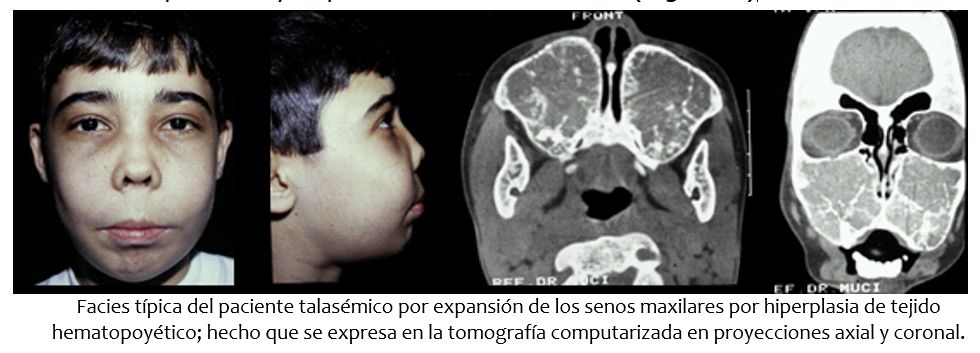
Con relación a la thalassemia, talasemia o beta-talasemia, descrita por Thomas Benton Cooley en 1925, se caracteriza por déficit en la síntesis de las cadenas ß de la hemoglobina. Su herencia es autosómica recesiva, y en mucho menor proporción, autosómica dominante. Los sufrientes presentan anemia crónica, hiperplasia compensadora del tejido hematopoyético: hepatoesplenomegalia, expansión de la médula ósea así que la cortical ósea se adelgaza lo que conduce a falta de neumatización de los senos maxilares y a fracturas patológicas; los huesos de la cara y el cráneo se deforman dando lugar a una facies peculiar –prominencia de los malares, de los incisivos superiores y separación de las órbitas- (Figura 6)
El tratamiento de referencia para la talasemia mayor son las transfusiones de sangre cada 2 a 4 semanas para mantener un nivel promedio de hemoglobina de 12.0 g/dL para controlar la anemia, suplir la oxigenación tisular, suprimir la producción de eritrocitos defectuosos y evitar los cambios óseos característicos craneales y faciales y la esplenomegalia y así, aumentar la expectativa de vida. Adicionalmente, debe complementarse con el tratamiento quelante o atrapador para evitar la sobrecarga de hierro, empleando desferoxamina parenteral (Desferal®), y más recientemente las drogas orales deferiprone-L1 (Ferriprox®) y el deferasirox (Exjade®).
Aunque carezco de ilustración en la materia, tal vez compense mi insuficiencia el tocarme el honor de la misión de comentar en general, el extenso, meduloso y valioso artículo del Doctor Guevara Iribarren. El autor introduce el tema comentando la evolución del conocimiento mundial en el campo de las hemoglobinas y el sucesivo descubrimiento y descripción de las principales variantes normales y anormales.
De su lectura se concluye que en Venezuela el campo de investigación de las hemoglobinas anormales, ha sido uno muy serio, activo y secuencial; en su trabajo analiza las más importantes investigaciones sobre hemoglobinas anormales realizadas en un período de 52 años, entre 1956-2009. El universo objeto de este estudio estuvo constituido por poblaciones mestizas normales, negroides, indígenas y en pacientes hospitalarios y sus familiares. Para facilitar el transitar por el progreso del conocimiento de estas enfermedades en Venezuela, el autor describe tres etapas.
La primera se refiere a las descripciones primigenias en el país mediante la identificación de drepanocitos en sangre periférica y en las cuales tuvieron injerencia destacados miembros de esta honorable Academia. Otto Lima Gómez y Luis Carbonell en 1946 (6), realizaron en Paparo, población del Estado Miranda, el primer estudio de incidencia de células falciformes en Venezuela. Analizaron los frotis sanguíneos de 40 individuos de raza negra con tinción de azul de cresil brillante, hallando una frecuencia de 5%. Un año después, en 1947, Henrique Benaím Pinto, Luis Carbonell, J. A. Gil y Otto Lima Gómez (7) publicaron la primera descripción de pacientes con anemia drepanocítica en Venezuela; y en 1950, H. Ossot y Oscar Agüero (8) informaron del primer caso en una mujer embarazada. En 1953, José Barnola y cols. (9,10), evaluaron más de seis mil conscriptos y tres mil niños obteniendo una frecuencia de positivos de 3,1% y 3,2% respectivamente. En 1961, Tulio Arends encontró una frecuencia de 5,2% en 210 personas analizadas (11).
En una segunda etapa se avanza al método electroforético y la metodología empleada fue cambiando al son de los adelantos tecnológicos en ese campo. Se analizó una cohorte superior a las 26 mil muestras procedentes de todo el país; la mayor frecuencia de variantes anormales de hemoglobinas, especialmente HbS y HbC, se encontró en anémicos y sus familiares y población negroide de origen africano. Hace 54 años, Tulio Arends, entre 1956 y 1962, determinó que la HbS en forma heterocigota, dependiendo de la mezcla étnica de las comunidades mostraba una frecuencia variable que iba desde un 19% en poblaciones formadas por descendientes de esclavos africanos hasta su ausencia en los estados andinos (Arends, 1971; Arends y cols. 1978). Demostró que, en las zonas de alta frecuencia, esta hemoglobinopatía constituía un problema de salud pública (Arends, 1984). Debe destacarse que con excepción de la persistencia hereditaria de la hemoglobina fetal (HPFH) tipo Venezuela, encontrada en indios de la etnia Warao del Delta del Orinoco, en el resto de la población indígena no se halló variantes hemoglobínicas. Se postuló así que toda variante con frecuencia mayor de 1% o superior, debía ser considerada como endémica y representante potencial de un problema de salud pública en la región.
La tercera etapa es marcada por la caracterización molecular. En 1994 la doctora Anabel Arends y cols., estudian más de ochenta mil muestras de sangre provenientes de diferentes regiones del país y de pacientes de los laboratorios de hemoglobinas anormales del Hospital Universitario de Caracas, UCV, Instituto Anatómico de la UCV y Hematología Experimental del IVIC; un grupo fue estudiado por electroforesis de hemoglobina en acetato de celulosa y citrato de agar, prueba de solubilidad, cuantificación de HbA2 por microcromatografía en columna y cuantificación de hemoglobina fetal por dos procedimientos diferentes. El 9% de estos sujetos presentaron hemoglobinopatías; la variante más frecuente de nuevo fue la HbS seguida de las variantes HbC y HbD. Además se observó la frecuencia de b talasemia y su asociación con las hemoglobinas S y C.
Entre otras inquietudes en el campo de la hematología, con mucho, la vida profesional del Doctor Guevara Iribarren ha estado ocupada con el problema del estudio y caracterización las hemoglobinopatías en Venezuela, y en sus conclusiones asienta que, (1). En Venezuela la distribución de pacientes con hemoglobinas anormales es heterogénea; (2). El método de cromatografía líquida de alta presión (HPLC-CE) es rápido, sensible y preciso; (3). La drepanocitosis es una enfermedad multicromosomal; (4). La deleción a-3.7 talasemia es la más frecuente en nuestro país. (5). Se evidenció que la mezcla racial en los Waraos ocurrió en los últimos treinta años. En ellos se detectó la persistencia hereditaria de hemoglobina fetal (HPFH) de un tipo completamente diferente de los tipos africanos y griego, y con características similares al denominado tipo suizo, aunque la distribución intracelular de HbF es totalmente diferente, por lo que se ha propuesto la denominación de tipo venezolano (6). Empleando las variantes hemoglobínicas como marcadores genéticos, se pudo establecer que la mayoría de la población venezolana es un híbrido de indígenas, españoles y esclavos africanos; pero con la posterior contribución de inmigrantes italianos, españoles y portugueses se ha hecho multinacional. (7) Las hemoglobinopatías representan un problema de salud pública y por tanto, la pesquisa neonatal debe ser obligatoria.
Honorables académicos, colegas invitados y amigos,
Comprendo muy bien –porque la razón autocrítica no me flaquea-, que mi fortaleza es pequeña y mi capacidad intelectual limitada, especialmente en esta área de las hemoglobinopatías, y que el honor y la confianza con la que me habéis investido, supera las fronteras de mis merecimientos, por lo que mi gratitud queda a perpetuidad, comprometida con el recipiendario y con ustedes.
Con gran simpatía y genuino júbilo, saludo al nuevo numerario Doctor José Guevara Iribarren y le hago llegar mis más emocionadas felicitaciones por habernos traído un enjundioso trabajo en la oportunidad de su merecido ascenso a numerario en la Academia Nacional de Medicina, hasta cuyos integrantes extiendo mis parabienes por incorporar a su seno su acabada personalidad científica, ética y moral.
Ocupe Usted por merecimiento y justicia, el Sillón XVII de Individuo de Número.
Muchas gracias por su amable atención.
Referencias
- Paton D. The conjunctival sign of sickle cell disease. Arch Ophthalmol. 1961;66:116-120.
- Paton D. The conjunctival sign of sickle cell disease. Further observations. Arch Ophthalmol. 1962;68:627-632.
- Fred CP. Diagnosis of sickle cell disease by ophthalmoscopy inspection of the conjunctiva. New Engl J Med. 1964;271:544-546.
- Muci-Mendoza R, Wuani H, Vegas H, Cruz A. Alteraciones oculares en la anemia drepanocítica con especial referencia a las conjuntivales. Arch Hosp Vargas. 1968;10:49-62.
- Welch R, Goldberg M. Sickle cell hemoglobin and its relation to fundus abnormalitiy. Arch Ophthalmol. 1966;75:353-362.
- Gómez OL, Carbonell L. Drepanocitos en Venezuela. S.E.M (Caracas).1946;13:18-23.
- Benaím H, Carbonell L, Gil JA, Gómez OL. Primera descripción de la anemia drepanocítica en Venezuela. Rev Polic Caracas.1947;26:1.
- Ossot H, Agüero O. Anemia drepanocítica en embarazada. Bol Mat Concepción Palacios 1950; 1: 6.
- Barnola J, Tovar- Escobar G, Potenza L. Enfermedad por Células falciformes. Arch Venez Puer Ped 1953;16:293.
- Barnola J, Quintero-Uzcátegui H. Enfermedad por células falciformes y su importancia en cirugía. Primer Congreso de Cirugía. Editorial Sucre, 1952.
- Arends T. El problema de las hemoglobinopatías en Venezuela. Rev San Asist Social.1961;26:61-68.




 Don José de Letamendi i de Manjarrés (1828-1897), Catedrático de Patología General de la Universidad de Madrid, siempre pidió que el médico, si quería comprender al hombre y en especial al hombre enfermo, tuviera una formación humanística y filosófica al lado de la técnica. Su famosa frase: «El médico que sólo de medicina sabe, ni de medicina sabe», resume toda su filosofía. La medicina antropológica de Viktor von Weizsäcker (1886-1957), demostró que los síntomas patológicos pueden ser símbolos de conflictos anímicos y que, hasta los pensamientos, pueden ser causa de enfermedad. La angustia existencial produce neurosis, las neurosis son traducidas por el organismo en trastornos funcionales, y éstos por un mecanismo de fijación, se transforman en enfermedades orgánicas. No hay que olvidar que cuando el médico llega a ver al paciente por esos síntomas, las raíces de la enfermedad están profundamente introducidas no sólo en la personalidad del paciente, sino en el cuerpo social donde éste se encuentra integrado, o mejor dicho en muchos casos no integrado.
Don José de Letamendi i de Manjarrés (1828-1897), Catedrático de Patología General de la Universidad de Madrid, siempre pidió que el médico, si quería comprender al hombre y en especial al hombre enfermo, tuviera una formación humanística y filosófica al lado de la técnica. Su famosa frase: «El médico que sólo de medicina sabe, ni de medicina sabe», resume toda su filosofía. La medicina antropológica de Viktor von Weizsäcker (1886-1957), demostró que los síntomas patológicos pueden ser símbolos de conflictos anímicos y que, hasta los pensamientos, pueden ser causa de enfermedad. La angustia existencial produce neurosis, las neurosis son traducidas por el organismo en trastornos funcionales, y éstos por un mecanismo de fijación, se transforman en enfermedades orgánicas. No hay que olvidar que cuando el médico llega a ver al paciente por esos síntomas, las raíces de la enfermedad están profundamente introducidas no sólo en la personalidad del paciente, sino en el cuerpo social donde éste se encuentra integrado, o mejor dicho en muchos casos no integrado.





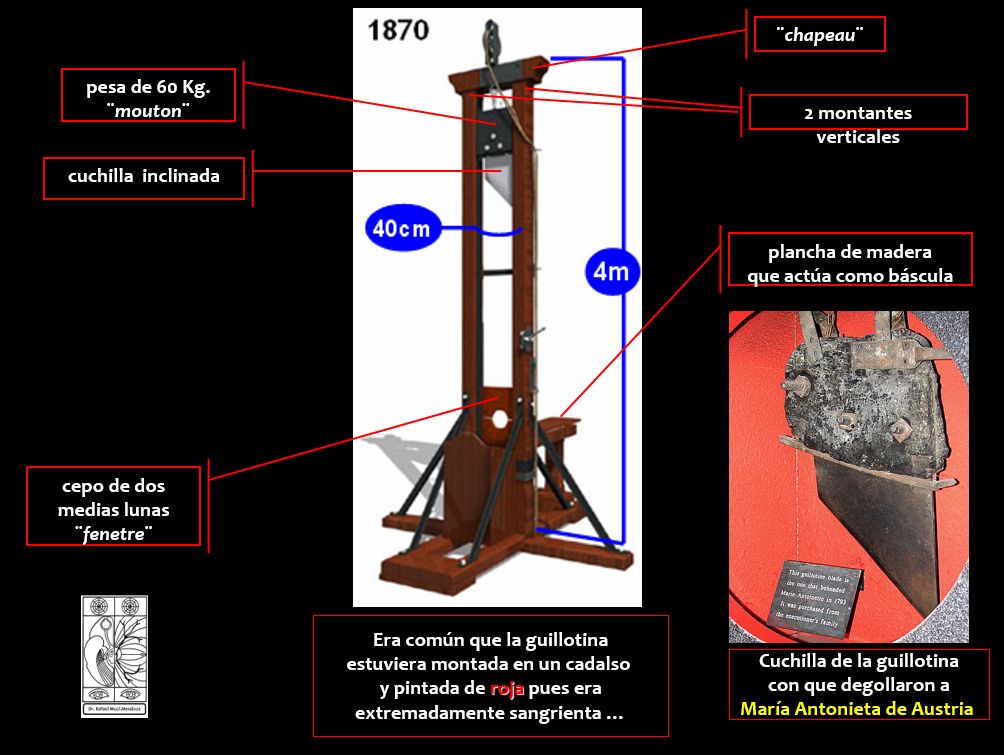











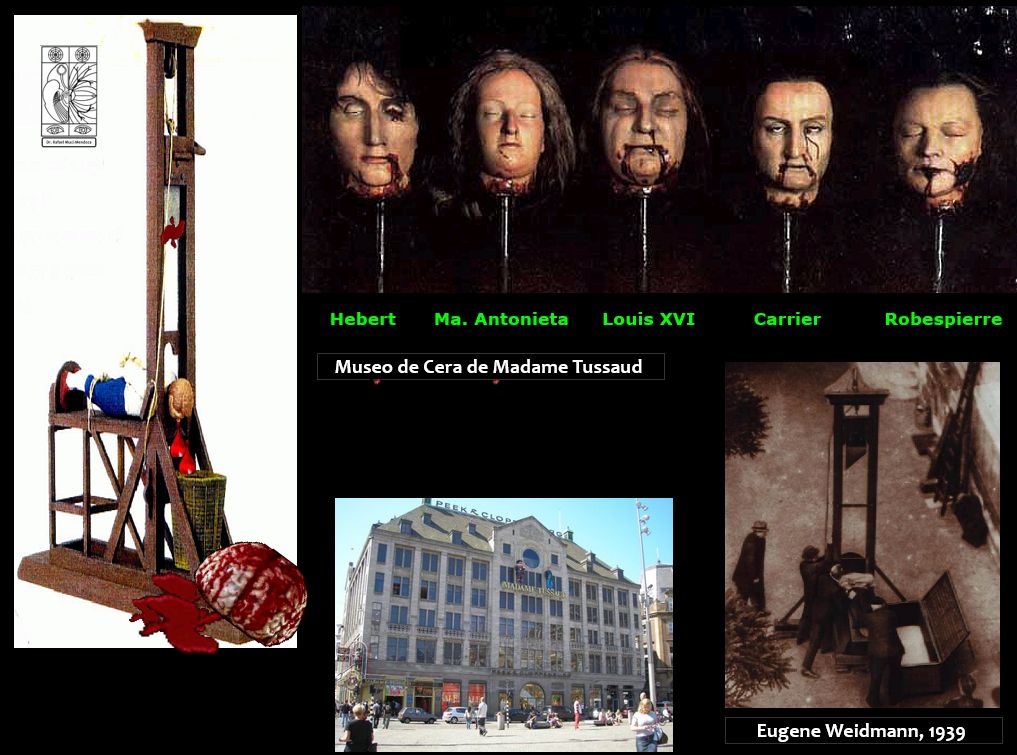
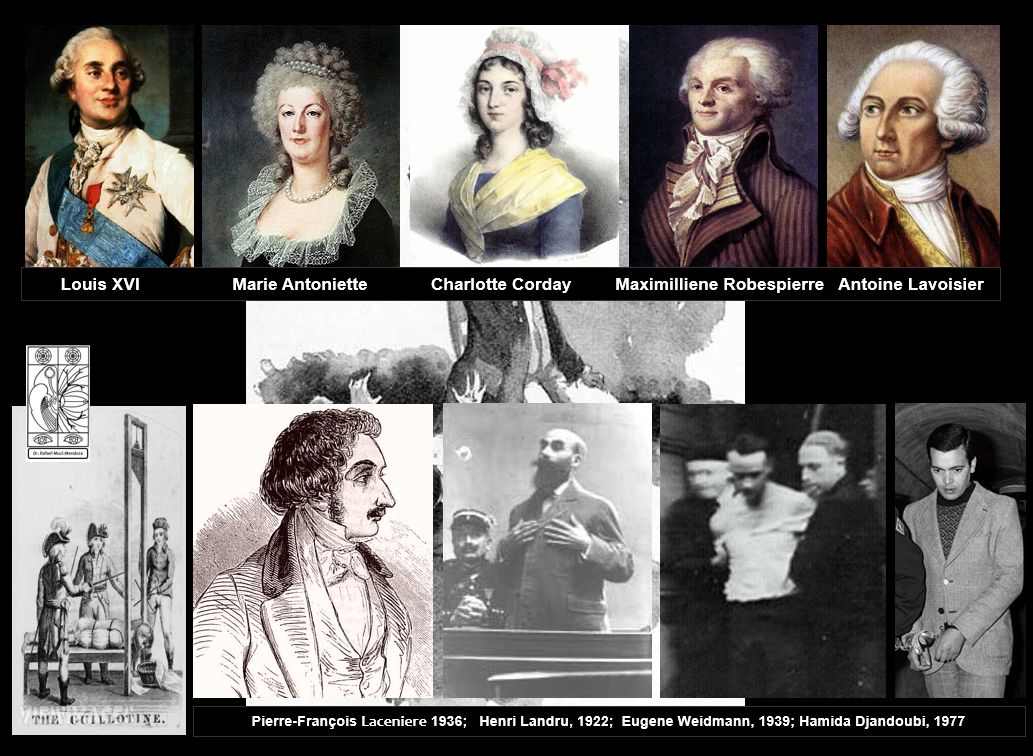






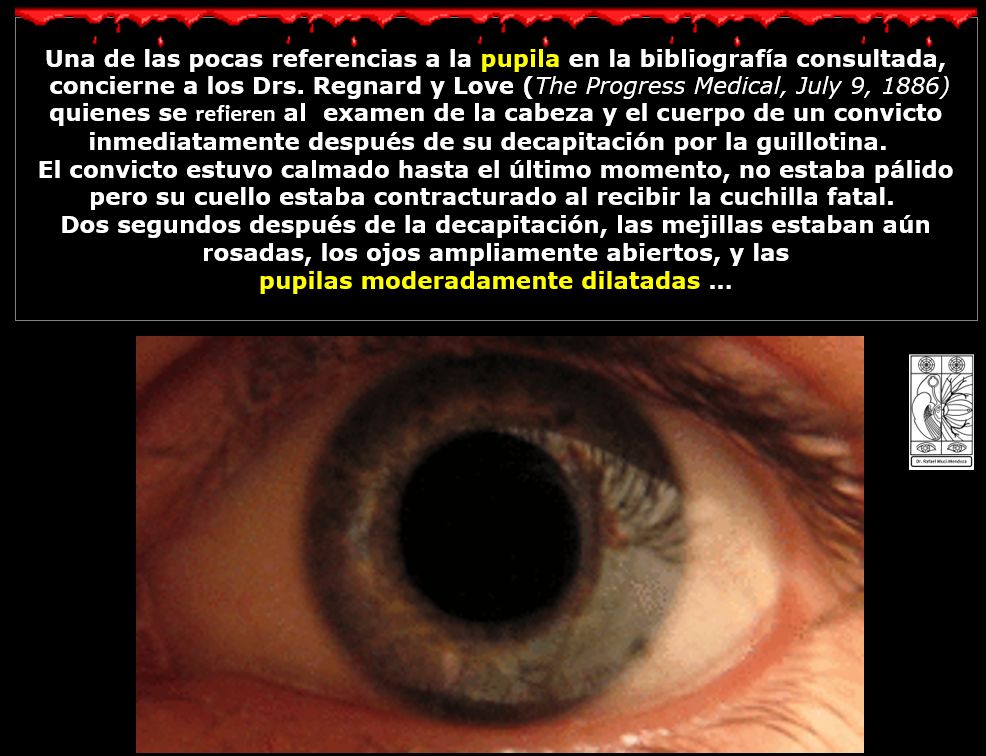

 Los médicos Piedlievre y Fournier asentaron que la muerte por la guillotina ¨no es instantánea realmente, cada elemento vital sobrevive a la decapitación… es una vivisección salvaje seguida de entierro prematuro¨ (12).
Los médicos Piedlievre y Fournier asentaron que la muerte por la guillotina ¨no es instantánea realmente, cada elemento vital sobrevive a la decapitación… es una vivisección salvaje seguida de entierro prematuro¨ (12).

 Siendo que la leyenda es notable, la verdadera historia es aún más asombrosa. Nuestro héroe comandaba una misión más dura e importante; el ejército persa se aprestaba a destruir a Atenas y él fue comisionado a ir a Esparta que distaba 240 kilómetros en búsqueda de ayuda; la trayectoria era demasiado accidentada para los caballos, así que no quedaba otra opción que enviar un emisario que, corriendo, cubriera el trayecto. Y así lo hizo en dos días, pero para su frustración, los espartanos celebraban el festival de Artemisa y no accedieron a prestarles asistencia; tuvo entonces que devolverse trotando similar cantidad de kilómetros a traer las malas nuevas, pero le dio fuerzas el ver durante la ruta al Dios Pan, dios de la fertilidad y de la sexualidad masculina desenfrenada, aunque se sospecha que fue una alucinación producto del extremo calor…
Siendo que la leyenda es notable, la verdadera historia es aún más asombrosa. Nuestro héroe comandaba una misión más dura e importante; el ejército persa se aprestaba a destruir a Atenas y él fue comisionado a ir a Esparta que distaba 240 kilómetros en búsqueda de ayuda; la trayectoria era demasiado accidentada para los caballos, así que no quedaba otra opción que enviar un emisario que, corriendo, cubriera el trayecto. Y así lo hizo en dos días, pero para su frustración, los espartanos celebraban el festival de Artemisa y no accedieron a prestarles asistencia; tuvo entonces que devolverse trotando similar cantidad de kilómetros a traer las malas nuevas, pero le dio fuerzas el ver durante la ruta al Dios Pan, dios de la fertilidad y de la sexualidad masculina desenfrenada, aunque se sospecha que fue una alucinación producto del extremo calor…

